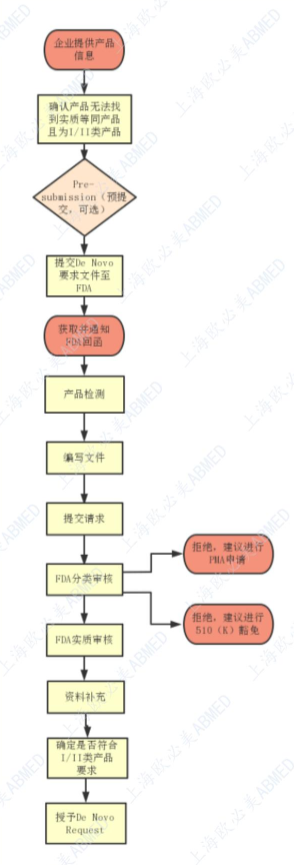
FDA De Novo分类请求的申请流程
De Novo路径提供了一种上市许可途径,适用于没有前代产品的器械分类,也不需要提交PMA流程的情形。通过审评相关证据,如发现该医疗器械的风险通过常规控制措施或者特殊控制措施即可保证医疗器械的安全有效性,则该医疗器械可被分类为I类或者II类(III类进行PMA申请)。
通过De Novo分类请求被分类为I类或II类的设备可被上市,并可用作未来510(k)提交的比对产品(如适用)。
在向FDA提交De Novo申请之前,FDA建议申请者先考虑进行预提交pre-sub,提前沟通其所需要提交的支持De novo 分类的额外信息,以促进整个流程更高效。
有两种选择可以向FDA提交De Novo请求,以便对分类为I类或II类中低风险器械进行基于风险的评估。
途径1:在收到针对510(k)提交的非实质性等同(NSE)确定(即,无比对器械、新的预期用途或引发不同安全性和有效性问题的不同技术特征)后。
途径2:确定没有合法销售的器械可作为实质等效性(SE)的确定依据。