
400-0569-812
400-0569-812



医疗器械制造商将受益于以下修订:
对MDR的修订:
★ 延长2021年5月26日之前根据医疗器械指令(MDD)或有源植入式器械指令(AIMDD)颁发的CE证书所涵盖的设备的过渡期:
1. III 类设备和大多数 IIb 类植入式设备适用至2027 年12 月 31 日(但缝合线、钉、 牙科填充物、牙套、牙冠、骨钉、骨楔、骨板、线、针、夾子和连接器除外);
2. 其他 IIb 类设备、IIa 类设备和 Is 或 Im 类设备适用至 2028年12月 31日。
★ 这些延长的过渡期仅在满足以下条件时才适用:
1. 该设备继续符合 MDD/AIMDD (具体指90/385/EEC 或指令 93/42/EEC);
2. 该设备的设计或预期用途未发生任何重大变化化(基于 Article 120 MDR 的定义);
3. 该设备不会对患者和用户的健康和安全构成不可接受的风险;
4. 截至 2024年 5 月 26 日,制造商已根据MDR第10(9)条建立了质量管理体系;
5. 截至2024年 5 月 26 日,制造商已根据MDR提交了正式的符合性评估申请,并在2024年9月26日之前与公告机构签署了关于MDR符合性评估的书面协议。
★ 如不满足这些要求,制造商就无法从延期中受益。
在签署 MDR 认证协议后,NB 根据 MDR 要求对 MDD/AIMDD 证书进行“适当的监督审核”。根据 MDD/AIMDD 证书的状态 “适当的监督审核” 于定期监督审核中结合文件审查、扩充审核等活动进行。
★ 将MDD/AIMDD证书延长至相应过渡期结束(如上所述)–这也适用于在修订生效之前已过期的证书,如果:
1. 制造商和指定机构已就证书所涵盖的设备合格评定签署了书面协议;
2. 国家主管机关已根据MDR第59条第1款批准适用的合格评定程序的减少,或已要求制造商根据MDR第97条第1款在一定时间内执行合格评定程序。
★ 为III类定制植入式设备引入新的过渡期,直到2026年5月26日——目前的过渡期不包括这些设备。如果制造商已根据MDR提交了正式的合格评定申请,并且在2024年9月26日之前与指定机构签署了根据MDR的合格评定的书面协议,则此过渡期适用。
★ 根据MDR的过渡期取消投放市场的设备的抛售日期(目前为2025年5月27日)–过渡期结束后,将这些设备投放市场将没有时间限制。
体外诊断法规(IVDR)的修正案
★ 取消销售日期(目前介于2025年5月26日至2028年5月26日之间,取决于IVDR中器械的风险级别)。
不延长过渡期。一般来说,根据IVDD颁发证书的设备可以在2025年5月26日之前投放市场。根据指令IVDD的符合性评估程序不要求公告机构参与的器械可以在2027年5月26日之前投放市场,这取决于器械的分类。
制造商所需操作
当修正案生效时,其MDD / AIMDD证书仍然有效的医疗设备制造商虽不必立即采取行动。但是,考虑到审核机构审核压力,制造商应尽快着手以下2点:
(i)联系指定机构并签署有关MDR合格评定的合同,这一点尤其重要,因为修正案不会解决公告机构缺乏能力的问题
(ii)建立MDR质量管理体系。
这两项行动都必须在 2024年 5 月 26日之前采取,才能从新的过渡规则中受益。
已经过期的MDD/AIMDD证书的制造商也可以根据MDR第97条申请程序,前提是他们满足医疗器械协调小组(MDCG)规定的要求。在特定情况下,根据MDR第57条申请减损也可能是一种选择,尽管根据MDCG,这一规定不应作为延迟MDR合格评估程序的解决方案。
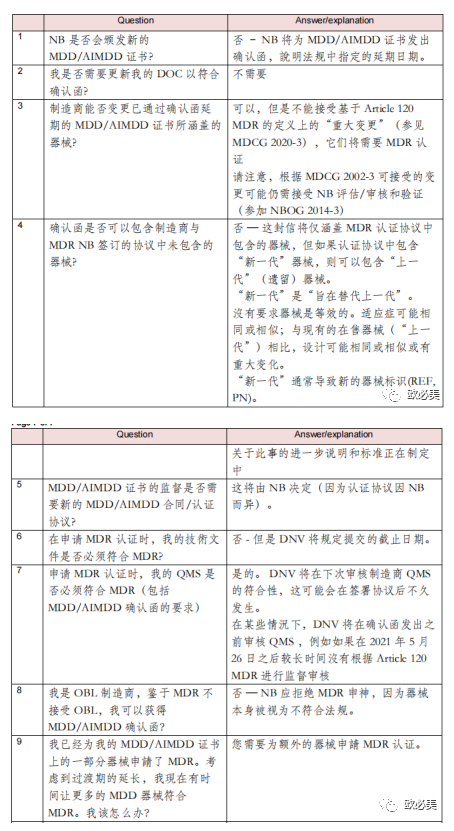
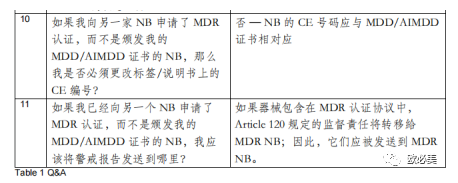
制造商常见问题
自2022 年 12 月公布延期修正案以來,大家提出了许多问题,大多和审核机构有关,以下我们以DNV的部分回复为例:
上海欧必美医疗技术集团有限公司 版权所有 Copyright© 2022-2026 All rights reserved 沪ICP备19022935号-1
