
400-0569-812
400-0569-812




FDA体外诊断和放射健康办公室主任蒂莫西·斯坦泽尔(Timothy Stenzel)在新冠肺炎(COVID-19)和猴痘测试开发商的虚拟市政厅会议上表示,FDA正在对已经提交授权的测试进行最后审查,预计只会提交少量额外测试。目前尚不清楚闭幕词是否也提到了猴痘EUA,机构官员没有立即回应澄清要求。
FDA官员在2022年9月的政策更新中表示,他们打算只审查一小部分新的EUA诊断测试请求,并鼓励所有类型体外诊断测试的开发人员通过De Novo或510(k)途径获取上市批准。
FDA官员表示,机构计划给予已获得EUA授权的厂商180天宽限期,到期后EUA授权将不再有效。
一旦紧急使用授权被终止,随之而来的是FDA结束由于新冠肺炎疫情期间签发的新冠病毒诊断试剂,监护设备和呼吸机等设备的EUA。
这个监管信号其实已经释放很久了,早在去年年初,FDA就发布了针对医疗器械制造商指南草案,包括新冠试剂盒检测产品开发商,指导他们如何从紧急使用授权过渡到全面的上市及监管许可,其中包括一个过渡实施计划。如果制造商打算为他们的测试寻求全面监管批准,FDA建议他们包括一个过渡实施计划,解决他们将如何处理已经分发的测试。对于已经获得EUA的IVD体外诊断产品,一旦紧急使用授权被终止,在终止日期前销售的IVD体外诊断产品必须在两年内使用完。监护设备和呼吸机等治疗产品:对于已经获得紧急使用授权的治疗设备,FDA要求相应的产品制造商必须申请FDA的上市流程,或者贴上该产品未经FDA批准的标签。FDA将留出足够的时间,用于和EUA厂商就相应产品的后续处理进行充分的协商。
目前,FDA已经批准了大约440个新冠的检测试剂盒和7个猴痘检测试剂盒EUA授权。
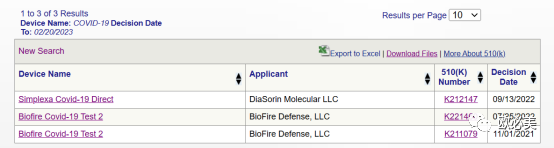
在FDA数据库查询,目前只有3家公司产品通过了FDA 510K的申请。
详情请咨询欧必美,我们用专业的知识为您解答。
上海欧必美医疗技术集团有限公司 版权所有 Copyright© 2022-2026 All rights reserved 沪ICP备19022935号-1
